 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChemical Reaction Prediction
Chemical reaction prediction is the process of predicting the outcome of chemical reactions using machine learning models.
Papers and Code
FragmentFlow: Scalable Transition State Generation for Large Molecules
Feb 02, 2026Transition states (TSs) are central to understanding and quantitatively predicting chemical reactivity and reaction mechanisms. Although traditional TS generation methods are computationally expensive, recent generative modeling approaches have enabled chemically meaningful TS prediction for relatively small molecules. However, these methods fail to generalize to practically relevant reaction substrates because of distribution shifts induced by increasing molecular sizes. Furthermore, TS geometries for larger molecules are not available at scale, making it infeasible to train generative models from scratch on such molecules. To address these challenges, we introduce FragmentFlow: a divide-and-conquer approach that trains a generative model to predict TS geometries for the reactive core atoms, which define the reaction mechanism. The full TS structure is then reconstructed by re-attaching substituent fragments to the predicted core. By operating on reactive cores, whose size and composition remain relatively invariant across molecular contexts, FragmentFlow mitigates distribution shifts in generative modeling. Evaluated on a new curated dataset of reactions involving reactants with up to 33 heavy atoms, FragmentFlow correctly identifies 90% of TSs while requiring 30% fewer saddle-point optimization steps than classical initialization schemes. These results point toward scalable TS generation for high-throughput reactivity studies.
MolecularIQ: Characterizing Chemical Reasoning Capabilities Through Symbolic Verification on Molecular Graphs
Jan 21, 2026A molecule's properties are fundamentally determined by its composition and structure encoded in its molecular graph. Thus, reasoning about molecular properties requires the ability to parse and understand the molecular graph. Large Language Models (LLMs) are increasingly applied to chemistry, tackling tasks such as molecular name conversion, captioning, text-guided generation, and property or reaction prediction. Most existing benchmarks emphasize general chemical knowledge, rely on literature or surrogate labels that risk leakage or bias, or reduce evaluation to multiple-choice questions. We introduce MolecularIQ, a molecular structure reasoning benchmark focused exclusively on symbolically verifiable tasks. MolecularIQ enables fine-grained evaluation of reasoning over molecular graphs and reveals capability patterns that localize model failures to specific tasks and molecular structures. This provides actionable insights into the strengths and limitations of current chemistry LLMs and guides the development of models that reason faithfully over molecular structure.
Generating readily synthesizable small molecule fluorophore scaffolds with reinforcement learning
Jan 12, 2026Developing new fluorophores for advanced imaging techniques requires exploring new chemical space. While generative AI approaches have shown promise in designing novel dye scaffolds, prior efforts often produced synthetically intractable candidates due to a lack of reaction constraints. Here, we developed SyntheFluor-RL, a generative AI model that employs known reaction libraries and molecular building blocks to create readily synthesizable fluorescent molecule scaffolds via reinforcement learning. To guide the generation of fluorophores, SyntheFluor-RL employs a scoring function built on multiple graph neural networks (GNNs) that predict key photophysical properties, including photoluminescence quantum yield, absorption, and emission wavelengths. These outputs are dynamically weighted and combined with a computed pi-conjugation score to prioritize candidates with desirable optical characteristics and synthetic feasibility. SyntheFluor-RL generated 11,590 candidate molecules, which were filtered to 19 structures predicted to possess dye-like properties. Of the 19 molecules, 14 were synthesized and 13 were experimentally confirmed. The top three were characterized, with the lead compound featuring a benzothiadiazole chromophore and exhibiting strong fluorescence (PLQY = 0.62), a large Stokes shift (97 nm), and a long excited-state lifetime (11.5 ns). These results demonstrate the effectiveness of SyntheFluor-RL in the identification of synthetically accessible fluorophores for further development.
High-Fidelity Modeling of Stochastic Chemical Dynamics on Complex Manifolds: A Multi-Scale SIREN-PINN Framework for the Curvature-Perturbed Ginzburg-Landau Equation
Jan 13, 2026The accurate identification and control of spatiotemporal chaos in reaction-diffusion systems remains a grand challenge in chemical engineering, particularly when the underlying catalytic surface possesses complex, unknown topography. In the \textit{Defect Turbulence} regime, system dynamics are governed by topological phase singularities (spiral waves) whose motion couples to manifold curvature via geometric pinning. Conventional Physics-Informed Neural Networks (PINNs) using ReLU or Tanh activations suffer from fundamental \textit{spectral bias}, failing to resolve high-frequency gradients and causing amplitude collapse or phase drift. We propose a Multi-Scale SIREN-PINN architecture leveraging periodic sinusoidal activations with frequency-diverse initialization, embedding the appropriate inductive bias for wave-like physics directly into the network structure. This enables simultaneous resolution of macroscopic wave envelopes and microscopic defect cores. Validated on the complex Ginzburg-Landau equation evolving on latent Riemannian manifolds, our architecture achieves relative state prediction error $ε_{L_2} \approx 1.92 \times 10^{-2}$, outperforming standard baselines by an order of magnitude while preserving topological invariants ($|ΔN_{defects}| < 1$). We solve the ill-posed \textit{inverse pinning problem}, reconstructing hidden Gaussian curvature fields solely from partial observations of chaotic wave dynamics (Pearson correlation $ρ= 0.965$). Training dynamics reveal a distinctive Spectral Phase Transition at epoch $\sim 2,100$, where cooperative minimization of physics and geometry losses drives the solver to Pareto-optimal solutions. This work establishes a new paradigm for Geometric Catalyst Design, offering a mesh-free, data-driven tool for identifying surface heterogeneity and engineering passive control strategies in turbulent chemical reactors.
ChemBART: A Pre-trained BART Model Assisting Organic Chemistry Analysis
Jan 06, 2026Recent advances in large language models (LLMs) have demonstrated transformative potential across diverse fields. While LLMs have been applied to molecular simplified molecular input line entry system (SMILES) in computer-aided synthesis planning (CASP), existing methodologies typically address single tasks, such as precursor prediction. We introduce ChemBART, a SMILES-based LLM pre-trained on chemical reactions, which enables a unified model for multiple downstream chemical tasks--achieving the paradigm of "one model, one pre-training, multiple tasks." By leveraging outputs from a mask-filling pre-training task on reaction expressions, ChemBART effectively solves a variety of chemical problems, including precursor/reagent generation, temperature-yield regression, molecular property classification, and optimizing the policy and value functions within a reinforcement learning framework, integrated with Monte Carlo tree search for multi-step synthesis route design. Unlike single-molecule pre-trained LLMs constrained to specific applications, ChemBART addresses broader chemical challenges and integrates them for comprehensive synthesis planning. Crucially, ChemBART-designed multi-step synthesis routes and reaction conditions directly inspired wet-lab validation, which confirmed shorter pathways with ~30% yield improvement over literature benchmarks. Our work validates the power of reaction-focused pre-training and showcases the broad utility of ChemBART in advancing the complete synthesis planning cycle.
A Pre-trained Reaction Embedding Descriptor Capturing Bond Transformation Patterns
Jan 07, 2026With the rise of data-driven reaction prediction models, effective reaction descriptors are crucial for bridging the gap between real-world chemistry and digital representations. However, general-purpose, reaction-wise descriptors remain scarce. This study introduces RXNEmb, a novel reaction-level descriptor derived from RXNGraphormer, a model pre-trained to distinguish real reactions from fictitious ones with erroneous bond changes, thereby learning intrinsic bond formation and cleavage patterns. We demonstrate its utility by data-driven re-clustering of the USPTO-50k dataset, yielding a classification that more directly reflects bond-change similarities than rule-based categories. Combined with dimensionality reduction, RXNEmb enables visualization of reaction space diversity. Furthermore, attention weight analysis reveals the model's focus on chemically critical sites, providing mechanistic insight. RXNEmb serves as a powerful, interpretable tool for reaction fingerprinting and analysis, paving the way for more data-centric approaches in reaction analysis and discovery.
Kinetic-Mamba: Mamba-Assisted Predictions of Stiff Chemical Kinetics
Dec 16, 2025Accurate chemical kinetics modeling is essential for combustion simulations, as it governs the evolution of complex reaction pathways and thermochemical states. In this work, we introduce Kinetic-Mamba, a Mamba-based neural operator framework that integrates the expressive power of neural operators with the efficient temporal modeling capabilities of Mamba architectures. The framework comprises three complementary models: (i) a standalone Mamba model that predicts the time evolution of thermochemical state variables from given initial conditions; (ii) a constrained Mamba model that enforces mass conservation while learning the state dynamics; and (iii) a regime-informed architecture employing two standalone Mamba models to capture dynamics across temperature-dependent regimes. We additionally develop a latent Kinetic-Mamba variant that evolves dynamics in a reduced latent space and reconstructs the full state on the physical manifold. We evaluate the accuracy and robustness of Kinetic-Mamba using both time-decomposition and recursive-prediction strategies. We further assess the extrapolation capabilities of the model on varied out-of-distribution datasets. Computational experiments on Syngas and GRI-Mech 3.0 reaction mechanisms demonstrate that our framework achieves high fidelity in predicting complex kinetic behavior using only the initial conditions of the state variables.
Learning Continuous Solvent Effects from Transient Flow Data: A Graph Neural Network Benchmark on Catechol Rearrangement
Dec 22, 2025

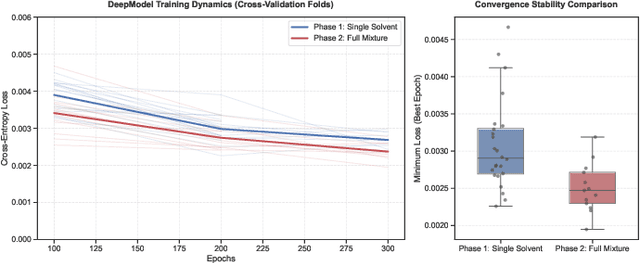

Predicting reaction outcomes across continuous solvent composition ranges remains a critical challenge in organic synthesis and process chemistry. Traditional machine learning approaches often treat solvent identity as a discrete categorical variable, which prevents systematic interpolation and extrapolation across the solvent space. This work introduces the \textbf{Catechol Benchmark}, a high-throughput transient flow chemistry dataset comprising 1,227 experimental yield measurements for the rearrangement of allyl-substituted catechol in 24 pure solvents and their binary mixtures, parameterized by continuous volume fractions ($\% B$). We evaluate various architectures under rigorous leave-one-solvent-out and leave-one-mixture-out protocols to test generalization to unseen chemical environments. Our results demonstrate that classical tabular methods (e.g., Gradient-Boosted Decision Trees) and large language model embeddings (e.g., Qwen-7B) struggle with quantitative precision, yielding Mean Squared Errors (MSE) of 0.099 and 0.129, respectively. In contrast, we propose a hybrid GNN-based architecture that integrates Graph Attention Networks (GATs) with Differential Reaction Fingerprints (DRFP) and learned mixture-aware solvent encodings. This approach achieves an \textbf{MSE of 0.0039} ($\pm$ 0.0003), representing a 60\% error reduction over competitive baselines and a $>25\times$ improvement over tabular ensembles. Ablation studies confirm that explicit molecular graph message-passing and continuous mixture encoding are essential for robust generalization. The complete dataset, evaluation protocols, and reference implementations are released to facilitate data-efficient reaction prediction and continuous solvent representation learning.
Learning continuous SOC-dependent thermal decomposition kinetics for Li-ion cathodes using KA-CRNNs
Dec 17, 2025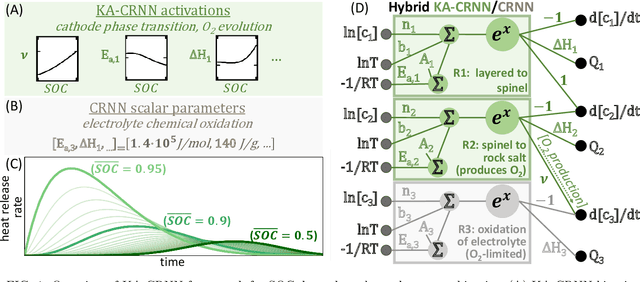
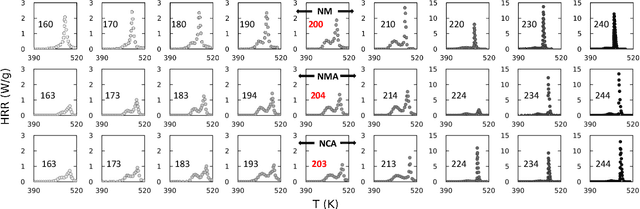
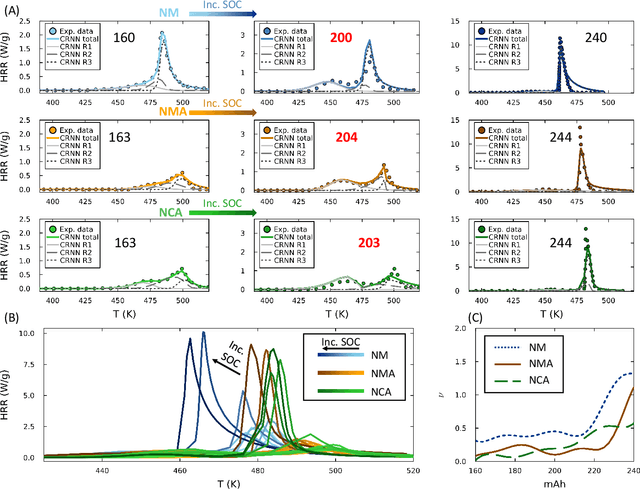
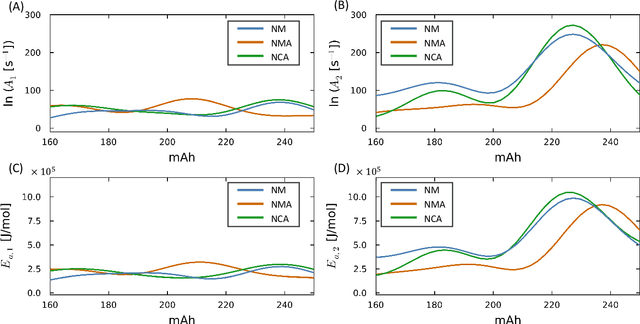
Thermal runaway in lithium-ion batteries is strongly influenced by the state of charge (SOC). Existing predictive models typically infer scalar kinetic parameters at a full SOC or a few discrete SOC levels, preventing them from capturing the continuous SOC dependence that governs exothermic behavior during abuse conditions. To address this, we apply the Kolmogorov-Arnold Chemical Reaction Neural Network (KA-CRNN) framework to learn continuous and realistic SOC-dependent exothermic cathode-electrolyte interactions. We apply a physics-encoded KA-CRNN to learn SOC-dependent kinetic parameters for cathode-electrolyte decomposition directly from differential scanning calorimetry (DSC) data. A mechanistically informed reaction pathway is embedded into the network architecture, enabling the activation energies, pre-exponential factors, enthalpies, and related parameters to be represented as continuous and fully interpretable functions of the SOC. The framework is demonstrated for NCA, NM, and NMA cathodes, yielding models that reproduce DSC heat-release features across all SOCs and provide interpretable insight into SOC-dependent oxygen-release and phase-transformation mechanisms. This approach establishes a foundation for extending kinetic parameter dependencies to additional environmental and electrochemical variables, supporting more accurate and interpretable thermal-runaway prediction and monitoring.
Accelerating High-Throughput Catalyst Screening by Direct Generation of Equilibrium Adsorption Structures
Dec 17, 2025The adsorption energy serves as a crucial descriptor for the large-scale screening of catalysts. Nevertheless, the limited distribution of training data for the extensively utilised machine learning interatomic potential (MLIP), predominantly sourced from near-equilibrium structures, results in unreliable adsorption structures and consequent adsorption energy predictions. In this context, we present DBCata, a deep generative model that integrates a periodic Brownian-bridge framework with an equivariant graph neural network to establish a low-dimensional transition manifold between unrelaxed and DFT-relaxed structures, without requiring explicit energy or force information. Upon training, DBCata effectively generates high-fidelity adsorption geometries, achieving an interatomic distance mean absolute error (DMAE) of 0.035 \textÅ on the Catalysis-Hub dataset, which is nearly three times superior to that of the current state-of-the-art machine learning potential models. Moreover, the corresponding DFT accuracy can be improved within 0.1 eV in 94\% of instances by identifying and refining anomalous predictions through a hybrid chemical-heuristic and self-supervised outlier detection approach. We demonstrate that the remarkable performance of DBCata facilitates accelerated high-throughput computational screening for efficient alloy catalysts in the oxygen reduction reaction, highlighting the potential of DBCata as a powerful tool for catalyst design and optimisation.